Assignment
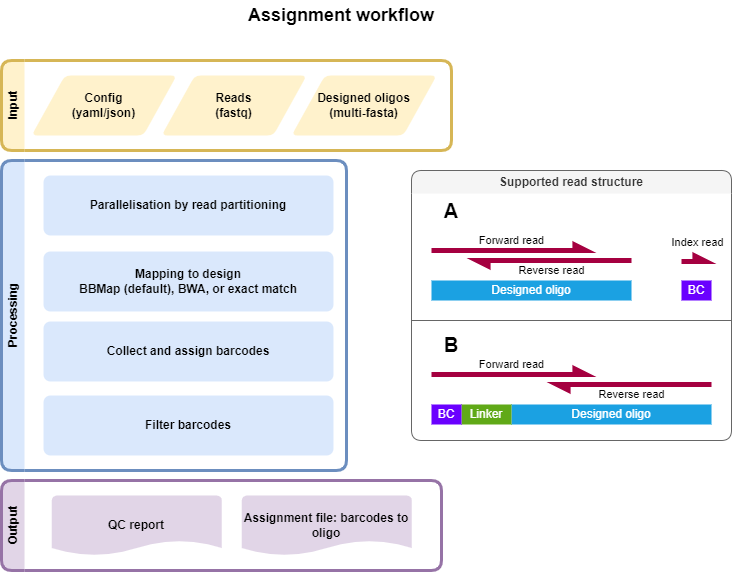
Input Files
Fastq Files
2-3 Fastq files from library association sequencing
Candidate regulatory sequence (CRS) sequencing, forward and reverse read (paired-end)
(optional) Index read with barcode. The barcode (BC) can also be present at the beginning of the forward read followed by a linker.
Design File
Multi-FASTA file of CRS sequences with unique headers describing each tested sequence.
Example file:
>CRS1
GACGGGAACGTTTGAGCGAGATCGAGGATAGGAGGAGCGGA
>CRS2
GGGCTCTCTTATATTAAGGGGGTGTGTGAACGCTCGCGATT
>CRS3
GGCGCGCTTTTTCGAAGAAACCCGCCGGAGAATATAAGGGA
>CRS4
TTAGACCGCCCTTTACCCCGAGAAAACTCAGCTACACACTC
Note
Headers of the design file must be unique (before any space), cannot contain [ or ], and should not contain duplicated sequences (forward and antisense).
Config File
Multiple mapping strategies are implemented to find the corresponding CRS sequence for each read. The mapping strategy can be chosen in the config file (bbmap, bwa mem, or exact matches). The config file also defines the filtering of the mapping results and is a YAML file.
Example of an assignment file using bbmap and the standard filtering (we recommend using bbmap as the default):
---
version: "0.5"
assignments:
exampleAssignment: # name of an example assignment (can be any string)
bc_length: 15
alignment_tool:
split_number: 1 # number of files fastq should be split for parallelization
tool: bbmap
configs:
min_mapping_quality: 30 # 30 is default for bbmap
sequence_length: 171 # sequence length of design excluding adapters.
alignment_start: 1 # start of an alignment in the reference/design_file. Here using 15 bp adapters. Can be different when using adapter free approaches
FW:
- resources/Assignment_BasiC/R1.fastq.gz
BC:
- resources/Assignment_BasiC/R2.fastq.gz
REV:
- resources/Assignment_BasiC/R3.fastq.gz
design_file: resources/design.fa
configs:
default: {} # name of an example filtering config
Example of an assignment file using bwa and the standard filtering:
---
version: "0.5"
assignments:
exampleAssignment: # name of an example assignment (can be any string)
bc_length: 15
alignment_tool:
split_number: 1 # number of files fastq should be split for parallelization
tool: bwa
configs:
min_mapping_quality: 1 # integer >=0 Please use 1 when you have oligos that differ by 1 base in your reference/design_file
sequence_length: # sequence length of design excluding adapters.
min: 166
max: 175
alignment_start: # start of an alignment in the reference/design_file. Here using 15 bp adapters. Can be different when using adapter free approaches
min: 1 # integer
max: 3 # integer
FW:
- resources/Assignment_BasiC/R1.fastq.gz
BC:
- resources/Assignment_BasiC/R2.fastq.gz
REV:
- resources/Assignment_BasiC/R3.fastq.gz
design_file: resources/design.fa
configs:
default: {} # name of an example filtering config
Example of an assignment file using exact matches with non-default filtering of barcodes:
---
version: "0.5"
assignments:
exampleAssignment: # name of an example assignment (can be any string)
bc_length: 15
alignment_tool:
split_number: 1 # number of files fastq should be split for parallelization
tool: exact # bwa or exact
configs:
sequence_length: 171 # sequence length of design excluding adapters.
alignment_start: 1 # start of the alignment in the reference/design_file
FW:
- resources/Assignment_BasiC/R1.fastq.gz
BC:
- resources/Assignment_BasiC/R2.fastq.gz
REV:
- resources/Assignment_BasiC/R3.fastq.gz
design_file: resources/design.fa
configs:
lazy: # name of an example filtering config
min_support: 2 # default 3
fraction: 0.6 # default 0.75
Example of an assignment file using exact matches and read 1 with BC, linker, and oligo (no separate BC index read):
---
version: "0.5"
assignments:
exampleAssignment: # name of an example assignment (can be any string)
bc_length: 20
BC_rev_comp: true
linker: TCTAGACCGTCACTAACTAACAGTGGGTACCC
alignment_tool:
split_number: 1 # number of files fastq should be split for parallelization
tool: exact # bwa or exact
configs:
sequence_length: 171 # sequence length of design excluding adapters.
alignment_start: 1 # start of the alignment in the reference/design_file
FW:
- resources/Assignment_BasiC/R1.fastq.gz
REV:
- resources/Assignment_BasiC/R3.fastq.gz
design_file: resources/design.fa
configs:
default: {} # name of an example filtering config
If you want to use the strand sensitivity option (e.g., testing enhancers in both directions), you can add the following to the config file: strand_sensitive: {enable: true}. Otherwise, MPRAsnakeflow will give you an error because it cannot handle the same sequences in both sense and antisense directions. This is an issue with the mappers because they do not consider the strand and will always call your read ambiguous due to multiple matches.
Snakemake
Options
With --help or -h, you can see the help message.
- Mandatory arguments:
- --cores:
Use at most N CPU cores/jobs in parallel. If N is omitted or ‘all’, the limit is set to the number of available CPU cores. In the case of cluster/cloud execution, this argument sets the number of total cores used over all jobs (made available to rules via workflow.cores). (default: None)
- --configfile:
Specify or overwrite the config file of the workflow (see the docs). Values specified in JSON or YAML format are available in the global config dictionary inside the workflow. Multiple files overwrite each other in the given order. Missing keys in previous config files are extended by following config files. (default: None)
- --sdm:
Required to run MPRAsnakeflow. :
--sdm condaor--sdm apptainer conda. Uses the defined conda environment per rule. We highly recommend using apptainer, where we build a predefined Docker container with all software installed within it.--sdm condainstalls the conda environments during the first execution of the workflow. If this flag is not set, the conda/apptainer directive is ignored. (default: False)
- Recommended arguments:
- --snakefile:
You should not need to specify this. By default, Snakemake will search for ‘Snakefile’, ‘snakefile’, ‘workflow/Snakefile’, or ‘workflow/snakefile’ beneath the current working directory, in this order. Only if you definitely want a different layout, you need to use this parameter. This is very useful when you want to have the results in a different folder than MPRAsnakeflow is in. (default: None)
- Useful arguments:
- -n:
Do not execute anything, and display what would be done. If you have a very large workflow, use –dry-run –quiet to just print a summary of the DAG of jobs. (default: False)
- --touch, -t:
Touch output files (mark them up to date without really changing them) instead of running their commands. This is used to pretend that the rules were executed, in order to fool future invocations of Snakemake. Fails if a file does not yet exist. Note that this will only touch files that would otherwise be recreated by Snakemake (e.g., because their input files are newer). For enforcing a touch, combine this with –force, –forceall, or –forcerun. Note, however, that you lose the provenance information when the files have been created in reality. Hence, this should be used only as a last resort. (default: False)
Rules
Rules run by Snakemake in the assignment utility:
all: The overall rule. Defines what final output files are expected.
all_assignments: Run all steps of the assignment workflow.
assignment_attach_idx: Extract the index sequence and add it to the header.
assignment_collect: Collect mapped reads into one BAM.
assignment_collectBCs: Get the barcodes.
assignment_fastq_split: Split the fastq files into N files for parallelization. N is given by split_read in the configuration file.
assignment_filter: Filter the barcodes file based on the config given in the config file. Results are here:
results/assignment/<assignment_name>/assignment_barcodes.<config_name>.tsv.gz.assignment_flagstat: Run samtools flagstat. Results are in
results/assignment/<assignment_name>/statistic/assignment/bam_stats.txt.assignment_mapping_bwa: Map the reads to the reference using BWA.
assignment_merge: Merge the forward, reverse, and barcode fastq files into one.
Output
The output can be found in the folder defined by the option results/assignment/. It is structured in folders of the condition as follows:
Files
Once the pipeline is finished running, all the output files can be seen in the results folder. This pipeline also generates a QC report.
For more details, refer to the HTML QC report.
File tree of the result folder (names in < > can be specified in the config file):
├── assignment
└── <assignment_name>
├── BCs
├── aligned_merged_reads.bam
├── aligned_merged_reads.bam.bai
├── assignment_barcodes.default.tsv.gz
├── assignment_barcodes_with_ambiguous.default.tsv.gz
├── barcodes_incl_other.tsv.gz
├── bbmap
├── design_check.done
├── design_check.err
├── fastq
│ └── splits
├── qc_report.default.html
├── reference
│ └── reference.fa
└── statistic
├── assigned_counts.default.tsv
├── assignment
│ └── bam_stats.txt
├── assignment.default.png
├── assignment.default.tsv.gz
└── total_counts.tsv
Key output files: - qc_report.default.html: QC report of the assignment. - total_counts.tsv: Raw statistics of barcodes mapped to oligos. - assigned_counts.<config_name>.tsv: Statistics of barcodes mapped to oligos after filtering. - assignment.<config_name>.tsv.gz: Average/median support of barcodes per oligo. - reference.fa: Design file. - aligned_merged_reads.bam: Sorted BAM file for oligo alignment. - assignment_barcodes.<config_name>.tsv.gz: Mapping file of barcodes to sequences.