Experiment (Count)
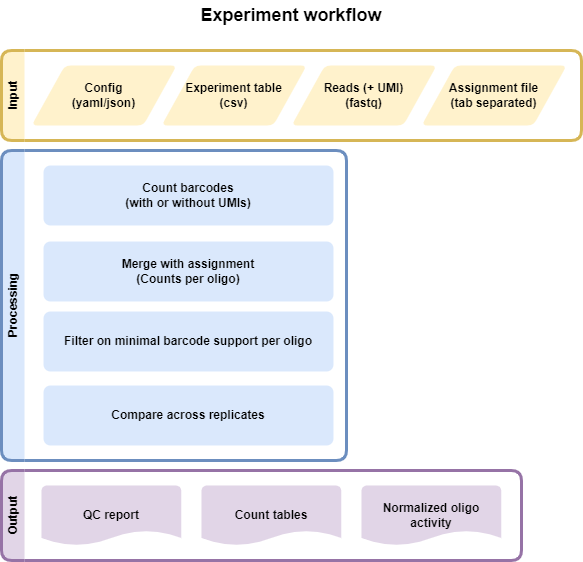
Input files
Experiment File
Comma separated file (CSV) that assigns all fastq files present in a directory to a condidtion and replicate. Each line represents an experiment, which will all be processed in parallel
Condition,Replicate,DNA_BC_F,DNA_UMI,DNA_BC_R,RNA_BC_F,RNA_UMI,RNA_BC_R
Condidtion1,1,C1R1_DNA_barcode_F.fastq.gz,C1R1_DNA_barcode_UMI.fastq.gz,C1R1_DNA_barcode_R.fastq.gz,C1R1_RNA_barcode_F.fastq.gz,C1R1_RNA_barcode_UMI.fastq.gz,C1R1_RNA_barcode_R.fastq.gz
Condidtion1,2,C1R2_DNA_barcode_F.fastq.gz,C1R2_DNA_barcode_UMI.fastq.gz,C1R2_DNA_barcode_R.fastq.gz,C1R2_RNA_barcode_F.fastq.gz,C1R2_RNA_barcode_UMI.fastq.gz,C1R2_RNA_barcode_R.fastq.gz
Condidtion1,3,C1R3_DNA_barcode_F.fastq.gz,C1R3_DNA_barcode_UMI.fastq.gz,C1R3_DNA_barcode_R.fastq.gz,C1R3_RNA_barcode_F.fastq.gz,C1R3_RNA_barcode_UMI.fastq.gz,C1R3_RNA_barcode_R.fastq.gz
Condidtion2,1,C2R1_DNA_barcode_F.fastq.gz,C2R1_DNA_barcode_UMI.fastq.gz,C2R1_DNA_barcode_R.fastq.gz,C2R1_RNA_barcode_F.fastq.gz,C2R1_RNA_barcode_UMI.fastq.gz,C2R1_RNA_barcode_R.fastq.gz
Condidtion2,2,C2R2_DNA_barcode_F.fastq.gz,C2R2_DNA_barcode_UMI.fastq.gz,C2R2_DNA_barcode_R.fastq.gz,C2R2_RNA_barcode_F.fastq.gz,C2R2_RNA_barcode_UMI.fastq.gz,C2R2_RNA_barcode_R.fastq.gz
Condidtion2,3,C2R3_DNA_barcode_F.fastq.gz,C2R3_DNA_barcode_UMI.fastq.gz,C2R3_DNA_barcode_R.fastq.gz,C2R3_RNA_barcode_F.fastq.gz,C2R3_RNA_barcode_UMI.fastq.gz,C2R3_RNA_barcode_R.fastq.gz
- We allow different flavours of experiment files because sometimes no UMI exists or only a FW read is used. Different options are:
Condition,Replicate,DNA_BC_F,DNA_UMI,DNA_BC_R,RNA_BC_F,RNA_UMI,RNA_BC_RCondition,Replicate,DNA_BC_F,DNA_BC_R,RNA_BC_F,RNA_BC_RCondition,Replicate,DNA_BC_F,RNA_BC_F
It is possible to use only one count experiment per condition across replicates (DNA or RNA, but usually only DNA can make sense). E.g. if you expect the same number of inserts/transfections across replicates. If you use the same files for DNA or RNA MPRAsnakeflow will only run the first replicate and use the counts for all replicates later.
Assignment File or configuration
Tab separated gzipped file with barcode mapped to sequence. Can be generated using the Assignment workflow. Config file must be configured similar to this:
example_assignment:
type: file
value: /path/to/your/file.tsv.gz
Example assignment file:
ATGCGT CRS1
GTCGA CRS2
CCGTT CRS3
CCCCT CRS4
Another option would be referring to an assignment defined in a config file.
example_assignment:
type: config
value: example_config
Label File (Optional)
Tab separated file (TSV) of desired labels for each tested sequence
Example file:
CRS1 Positive_Control
CRS2 Negative_Control
CRS3 Test
CRS4 Positive_Control
Note
If you provide a label file, the first column of the label file must exactly match the FASTA file or the files will not merge properly in the pipeline.
snakemake
Options
With --help or -h you can see the help message.
- Mandatory arguments:
- --cores:
Use at most N CPU cores/jobs in parallel. If N is omitted or ‘all’, the limit is set to the number of available CPU cores. In case of cluster/cloud execution, this argument sets the number of total cores used over all jobs (made available to rules via workflow.cores).(default: None)
- --configfile:
Specify or overwrite the config file of the workflow (see the docs). Values specified in JSON or YAML format are available in the global config dictionary inside the workflow. Multiple files overwrite each other in the given order. Thereby missing keys in previous config files are extended by following configfiles. Note that this order also includes a config file defined in the workflow definition itself (which will come first). (default: None)
- --sdm:
Required to run MPRAsnakeflow. :
--sdm condaor--sdm apptainer condaUses the defined conda environment per rule. We highly recommend to use apptainer where we build a predefined docker container with all software installewd within it.--sdm condathe conda envs will be installed by the first excecution of the workflow. If this flag is not set, the conda/apptainer directive is ignored. (default: False)
- Recommended arguments:
- --snakefile:
You should not need to specify this. By default, Snakemake will search for ‘Snakefile’, ‘snakefile’, ‘workflow/Snakefile’,’workflow/snakefile’ beneath the current working directory, in this order. Only if you definitely want a different layout, you need to use this parameter. This is very usefull when you want to have the results in a different folder than MPRAsnakeflow is in. (default: None)
- Usefull arguments:
- -n:
Do not execute anything, and display what would be done. If you have a very large workflow, use –dry-run –quiet to just print a summary of the DAG of jobs. (default: False)
- --touch, -t:
Touch output files (mark them up to date without really changing them) instead of running their commands. This is used to pretend that the rules were executed, in order to fool future invocations of snakemake. Fails if a file does not yet exist. Note that this will only touch files that would otherwise be recreated by Snakemake (e.g. because their input files are newer). For enforcing a touch, combine this with –force, –forceall, or –forcerun. Note however that you loose the provenance information when the files have been created in realitiy. Hence, this should be used only as a last resort. (default: False)
Rules
Rules run by snakemake in the experiment workflow. Some rules will be run only if certain options
- experiment_assigned_counts_assignBarcodes
Assign RNA and DNA barcodes seperately to make the statistic for assigned
- experiment_assigned_counts_combine_replicates
Combine replicates of master table by summing counts up and using also the average.
- experiment_assigned_counts_combine_replicates_barcode_output
Combine replictes of assigned barcode counts into one file.”””
- experiment_counts_umi_create_BAM
Create a BAM file from FASTQ input, merge FW and REV read and save UMI in XI flag.
- experiment_assigned_counts_copy_final_all_files
Will copy final files to the main folder so that it is creal which files to use.
- experiment_assigned_counts_copy_final_thresh_files
Will copy final files to the main folder so that it is creal which files to use.
- experiment_assigned_counts_dna_rna_merge
Assign merged RNA/DNA barcodes. Filter BC depending on the min_counts option.
- experiment_assigned_counts_filterAssignment
Use only unique assignments and do sampling if needed.
- experiment_assigned_counts_make_master_tables
Final master table with all replicates combined. With and without threshold.
- experiment_counts_demultiplex_BAM_umi
Demultiplexing the data and create demultiplexed bam files per condition.
- experiment_counts_demultiplex_aggregate
Aggregate the demultiplexed bam files per condition.
- experiment_counts_demultiplex_create_index
Create the demultiplexing index file for the experiment.
- experiment_counts_demultiplex_mergeTrimReads_BAM_umi
Merge and trim reads in demultiplexed bam files.
- experiment_counts_dna_rna_merge_counts
Merge DNA and RNA counts together. Is done in two ways. First no not allow zeros in DNA or RNA BCs (RNA and DNA min_counts not zero). Second with zeros, so a BC can be defined only in the DNA or RNA (RNA or DNA min_counts zero)
- experiment_counts_filter_counts
Filter the counts to BCs only of the correct length (defined in the config file)
- experiment_counts_final_counts
Counting BCs. Discarding PCR duplicates (taking BCxUMI only one time)
- experiment_counts_final_counts_sampler
Creates full + new distribution DNA files
- experiment_counts_noUMI_create_BAM
Create a BAM file from FASTQ input, merge FW and REV read and save UMI in XI flag.
- experiment_counts_noUMI_raw_counts
Counting BCsxUMIs from the BAM files.
- experiment_counts_onlyFWUMI_raw_counts
Getting the BCs and UMIs from the reads using fixed length.
- experiment_counts_onlyFW_raw_counts_by_cutadapt
Getting the BCs from the reads using cutadapt.
- experiment_counts_onlyFW_raw_counts_by_length
Getting the BCs from the reads using fixed length.
- experiment_counts_umi_raw_counts
Counting BCsxUMIs from the BAM files.
- experiment_statistic_assigned_counts_combine_BC_assignment_stats
Combined assinged counts statistic per condition (DNA and aRNA not merged)
- experiment_statistic_assigned_counts_combine_BC_assignment_stats_helper
Combine assigned counts statistic per replicate and modality (DNA and RNA not merged)
- experiment_statistic_assigned_counts_combine_stats_dna_rna_merge
Combine assigned counts statistic per replicate (DNA and RNA merged)
- experiment_statistic_assigned_counts_combine_stats_dna_rna_merge_all
Combine assigned counts statistic per condition (DNA and RNA merged)
- experiment_statistic_bc_overlap_combine_assigned_counts
Combine overlap BC and count statistic into one file (assigned counts).
- experiment_statistic_bc_overlap_combine_counts
Combine overlap BC and count statistic into one file (raw counts).
- experiment_statistic_bc_overlap_run
Get overlap of counts and barcodes between replicates.
- experiment_statistic_correlation_bc_counts
Calculate the correlation of the raw counts for each condition across replicates.
- experiment_statistic_correlation_bc_counts_hist
Generate histogram and boxplots of the raw counts for each condition across replicates.
- experiment_statistic_correlation_calculate
Calculate the correlation of oligos for each condition across replicates.
- experiment_statistic_correlation_combine_bc_assigned
Combine the correlation of the assigned counts for each condition across replicates into one table.
- experiment_statistic_correlation_combine_bc_raw
Combine the correlation of the raw counts for each condition across replicates into one table.
- experiment_statistic_correlation_combine_oligo
Combine the correlation of oligos for each condition across replicates into one table.
- experiment_statistic_correlation_hist_box_plots
Generate histogram and boxplots of the oligos for each condition across replicates.
- experiment_statistic_counts_BC_in_RNA_DNA
Count the number of barcodes shared between RNA and DNA per condition and replicate.
- experiment_statistic_counts_BC_in_RNA_DNA_merge
Merge the shared barcodes statistic of all replicates and conditions into one table.
- experiment_statistic_counts_barcode_base_composition
Count the nucleotide composition of the barcodes per condition, replicate and DNA/RNA.
- experiment_statistic_counts_final
Combine the final count statistic of all replicates and conditions into one table.
- experiment_statistic_counts_frequent_umis
Count the 10 most frequent UMIs per condition, replicate and DNA/RNA.
- experiment_statistic_counts_stats_merge
Merge the count statistic of all replicates and conditions into one table.
- experiment_statistic_counts_table
Count statistic of barcodes and UMIs per condition, replicate and DNA/RNA.
- experiment_statistic_quality_metric
Quality metrics of the assignment run
Output
The output can be found in the folder defined by the option results/experiments/. It is structured in folders of the condition as
Files
Once the pipline is finished running then all the output files can be seen in the results folder. This pipline also generates a qc report. For more details, refer to the HTML QC report.
File tree
experimet_name
|-Condition
|-allreps.tsv
|-average_allreps.tsv
|-HepG2_1_2_correlation.txt
|-HepG2_1_2_DNA_pairwise.png
|-HepG2_1_2_Ratio_pairwise.png
|-HepG2_1_2_RNA_pairwise.png
|-HepG2_barcodesPerInsert.png
|-Reps
|-HepG2_1_counts.tsv
|-HepG2_1_counts.tsv.gz
|-HepG2_1_DNA_counts_full.tsv
|-HepG2_1_DNA_counts_full_samplingN.tsv
|-HepG2_1_DNA_raw_counts.tsv.gz
|-HepG2_1_RNA_filtered_counts.tsv.gz
|-HepG2_1_DNA_filtered_counts.tsv.gz
|-HepG2_1_RNA_counts.tsv
|-HepG2_1_RNA_raw_counts.tsv.gz
Todo
This is not the correct file tree for the experiment workflow
Files for each Condition
- allreps.tsv
TSV of normalized DNA and RNA count, ratio, log2ratio, and number of observed barcodes for each condition, replicate, of every CRS
- average_allreps.tsv
mean ratio, log2 ratio, and observed barcodes per condidition normalized for all replicates
- HepG2_1_2_correlation.txt
correlation values for a condition and 2 replicates (ie: HepG2 replicate 1 vs replicate 2)
- HepG2_1_2_DNA_pairwise.png
Correlation plot of DNA counts condition vs two reps (ie: HepG2 replicate 1 vs replicate 2)
- HepG2_1_2_Ratio_pairwise.png
Correlation plot of normalized log2(RNA/DNA) condition vs two reps (ie: HepG2 replicate 1 vs replicate 2)
- HepG2_1_2_RNA_pairwise.png
Correlation plot of RNA counts condition vs two reps (ie: HepG2 replicate 1 vs replicate 2)
- HepG2_barcodesPerInsert.png
Histogram of number of barcodes detected per CRS
- HepG2_group_barcodesPerInsert_box.png
Boxplot of CRS normalized per insert, grouped by labels
Todo
These are not the correct files for each condition in the experiment workflow
Files for each replicate in each condition
- HepG2_1_counts.tsv
mean ratio, log2 ratio, and observed barcodes per condidition for each replicate
- HepG2_1_counts.tsv.gz
table of barcodes with DNA counts and RNA counts
- HepG2_1_DNA_counts_full.tsv
table of barcodes with DNA counts
- HepG2_1_DNA_counts_full_samplingN.tsv
table of barcodes with DNA counts with adjusted sampling.
- HepG2_1_DNA_raw_counts.tsv.gz
table of barcodes, UMI, and DNA counts raw
- HepG2_1_DNA_filtered_counts.tsv.gz
table of barcodes, UMI, and DNA counts raw, filtered for barcodes of correct length
- HepG2_1_RNA_counts.tsv
table of barcodes with RNA counts
- HepG2_1_RNA_raw_counts.tsv.gz
table of barcodes, UMI, and RNA counts raw
- HepG2_1_RNA_filtered_counts.tsv.gz
table of barcodes, UMI, and DNA counts raw, filtered for barcodes of correct length
Todo
These are not the correct files for the experiment workflow